Overview
Sickle cell disease is caused by a point mutation in the beta-globin gene found in chromosome 11. It results in a substitution of glutamine to valine at position 6, leading to the formation of the abnormal hemoglobin S. Sickle cell disease can be suspected in symptomatic children with anaemia, leukocytosis and thrombocytosis. There is no safe, effective, and curative treatment. The goal of treatment is mainly symptom control and management of disease complications.
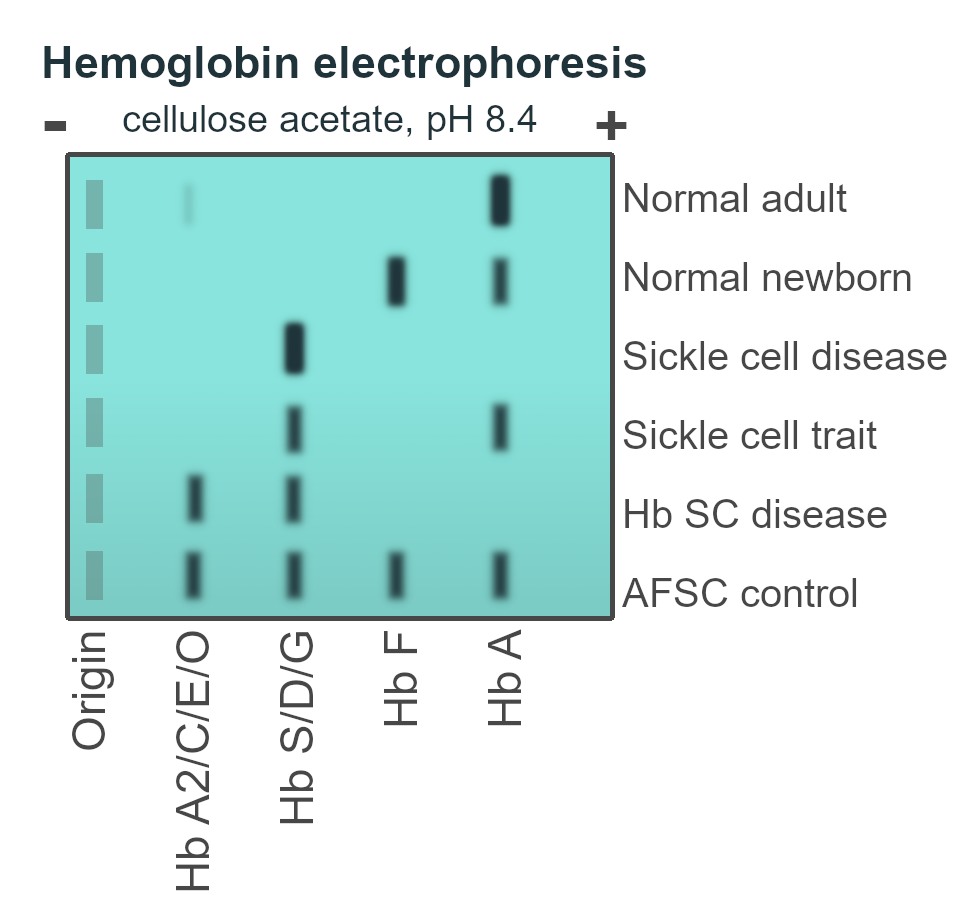
Sickle cell anemia: Homozygous (HbSS). Described in 1910. No HbA.
Sickle cell diseases: 2 defective genes. HbSS + HbSC/HbSD/HbSOarab/HbSE. No HbA.
Sickle cell trait: HbAS. Generally asymptomatic.
The carrier rate of HbS is between 2-40% in Africa. In western Kenya the carrier rate is 25%, with a 16% chance of both partners having HbAS. The chance of HbSS birth in western Kenya is 1 in 64. The incidence of stroke is highest between 2 – 9 years of age. 10% of children < 20 years get stroke. 25% get silent strokes.
Hemoglobin pattern
| Normal | Sickle Cell Disease | |
|---|---|---|
| HbA | 98% | None |
| HbF | 1% | 5-20% |
| HbA2 | <3.5% | 5% |
| HbS | None | 80% |
Lifespan of erythrocytes
| Erythrocyte | Days |
|---|---|
| Normal cells | 120 days |
| Sickle cells | 10 – 15 days |
Hemoglobin level
| Hemoglobin | Normal range |
|---|---|
| Normal | 14-16 g/dL |
| Sickle Cell Disease | 6-9 g/dL (despite active bone marrow) |

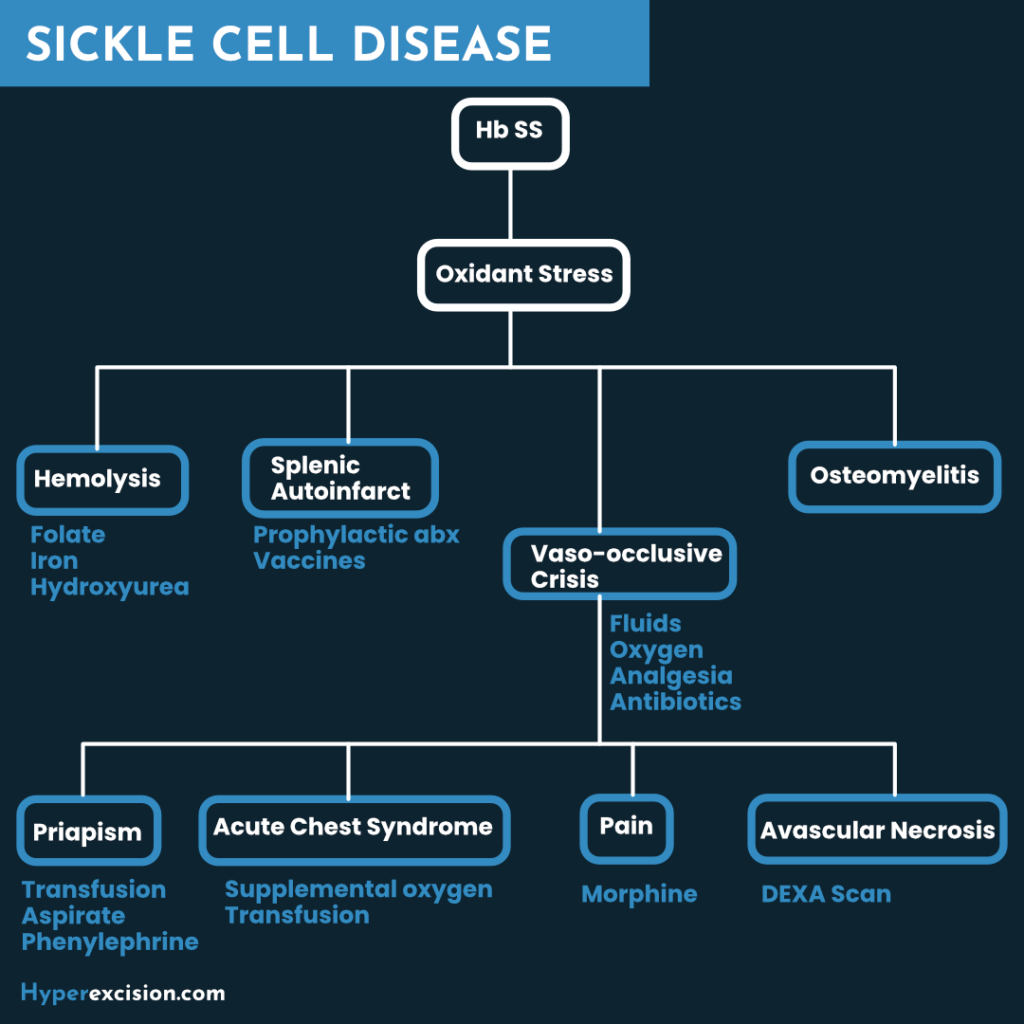
- Pathophysiology
- Hemoglobin S is soluble in the RBC as long as the cell is oxygenated.
- Hemoglobin S is unstable in hypoxia, dehydration, and acidosis
- Sickling begins when Oxygens saturation drop <85%. In heterozygotes the saturation have to be <40%.
- Hemoglobin molecules crystallize causing erythrocytes to become deformed (elongated, cigar shaped, sickle shaped)
- The deformed erythrocytes are less flexible in narrow vessels
- They can cause vaso-occlusion and are removed from circulation by the reticulo-endothelial system
- Compromised circulation causes end-organ pain, dysfunction, and necrosis
Clinical Presentation
Multisystem effects of SCD
| System | Effects (complications) |
|---|---|
| Lungs | ACS, Pulmonary fibrosis, Pulmonary HTN, Hypoxemia |
| CNS | Stroke, Subarachnoid hemorrhage, Intracerebral hemorrhage, Seizures, Cognitive impairment |
| GU | Priapism, Hematuria, papillary necrosis, glomerulonephritis |
| Eyes | Retinal artery occlusion, proliferative nephropathy, vitreous hemorrhage, retinal detachment |
| Immune system | Septicemia, Meningitis |
| MSK | Dactylitis, Avascular necrosis, Osteomyelitis |
| Skin | Chronic (leg) ulcers |
- When does sickle cell disease manifest?
- Manifests in early childhood
- For the first 6 months of life infants are protected by HbF
- After 6 months the condition becomes apparent.
- Factors that predict poor prognosis in children
- Dactylitis in infants < 12 months
- Hb level <7g/dl
- Leukocytosis in the absence of infection
- Signs and symptoms
- Pallor, Fatigue, and Shortness of breath (due to anaemia)
- Jaundice
- Pain
- Unpredictable Pains affecting any organ or joint
- Unpredicatble
- Start with hand/feet in young children
- Often of long bones, lower back, chest, and/or abdomen
- Specifically localized or migratory
- Variable quality (sharp, dull, stabbing, throbbing)
- Leg ulcers (chronic)
- Transient Ischemic Attacks and Stroke
- Infarction (most common): Hemiparesis, Altered speech and vision
- Hemorrhagic (common in adults): severe headache, vomiting, seizures, altered consciousness
- Fever and high-risk of infection (as the spleen becomes non-functional)
- Acute Chest Syndrome
- Pleuritic chest pain, Tachypnea, and Fever
- New pulmonary infiltrate
- Delayed growth and Delayed puberty
- Why do patients with sickle cell disease have a higher risk of infections
- Because of Autosplenectomy → defective opsonization
- Emergencies in Sickle Cell Disease
- Acute Chest Syndrome
- Aplastic Crisis
- Fever
- Multi-organ system failure
- Priapism
- Pain Crises
- Pulmonary Embolism
- Splenic sequestration
- Stroke
Management of SCD
- Investigations The diagnosis of sickle cell disease can be made on cord blood
- Complete Blood Count with reticulocyte count
- Low Hb
- High WBC
- High Platelet Count
- Reticulocytosis
- Peripheral Blood Film
- Sickle cell, Pencil Cells, Nucleated RBCs
- Sickling Test: confirmatory diagnosis where Hb electrophoresis is unavailable.
- Positive test in Sickle cell trait and Sickle cell disease
- Hemoglobin Electrophoresis: ****gold standard. Done 2-3 months after transfusion.
- Comprehensive metabolic panel
- CXR, Blood Cultures and Urinalysis: to rule out infection
- CT, MRI, or MRA: to confirm stroke
- Complete Blood Count with reticulocyte count
- Treatment goals in sickle cell disease
- Manage vaso-occlusive crisis
- Manage chronic pain syndrome
- Manage chronic hemolytic anemia
- Prevent and treat infections
- Manage complications and organ damage syndromes associated with the disease
- Prevent stroke
- Detect and treat pulmonary hypertension
- Prevention of Infection in SCD
- Penicillin prophylaxis from 4 – 6 months upto 5 years/early teens
- Penicillin V 125 mg PO BD until 2 years
- Penicillin V 250 mg PO BD from 2 years to 5 years
- 10 valent Pneumococcal vaccine at 6, 10, and 14 weeks
- 23 valent or 32 valent Pneumococcal Vaccine at age 2 years with booster at 5 years
- Does not give sero-conversion for children < 2 years of age
- Treat infections (fever + leukocytosis) with broad spectrum antibiotics promptly
- Penicillin prophylaxis from 4 – 6 months upto 5 years/early teens
- Treatment of Vaso-occulsive crisis
- Admit
- Oxygen
- Hydration (one and a half times the volume you would use)
- IV NS and D5
- Analgesics
- Paracetamol + ibruprofen at home
- Morphine in-patient
- NSAIDs (use with care due to nephrotoxicity)
- Broad spectrum antibiotics if there is an infection
- Antiemetics if there is nausea
- Antihistamines if there is pruritus
- Hydroxyurea to prevent chronic or recurrent episodes of pain
- Treatment of Anaemia
- Folic acid supplementation (supports a healthy reticulocyte count)
- Iron supplementations for menstruating women with co-existing iron deficiency
- Adequate diet
- Indications for blood transfusion in SCD to keep HbS <30% hence reducing sickling
- Splenic sequestration crisis
- Aplastic crisis
- Acute chest syndrome
- Stroke
- Stroke prevention in case of abnormal transcranial doppler
- Pregnancy
- General Anaesthesia
- What are the complications of transfusion in SCD?
- Alloimmunization (more chance of graft-vs-host reaction when looking for a stem cell donor)
- Iron overload (Subcutaneous iron chelation therapy is uncomfortable…)
- Increased risk of transfusion-transmitted infectious organisms
- What is the role of hydroxyurea in SCD? Comes in 500mg capsules. Long-term hydroxyurea is safe, even when started in young children.
- Increases the production of HbF reducing the frequency of sickling and vaso-occlusive crisis
- Reduces circulating leukocytes hence reducing adherence of neutrophils to the endothelium (WBC count should not be less than 2)
- Overall effect:
- Reduces chronic pain
- Reduces hospitalization
- Reduces Acute Chest Syndrome
- Reduces the need for transfusion
- Prevents early agieng of organs
- Decreases mortality
- What is the role of Bone Marrow Transplant in SCD?
- Allogenic BMT can cure SCD
- However, it is not 100% safe, and is expensive
- Cord Blood Stem Cell Transplant is safer than Bone Marrow Transplant
- Surgical management of SCD patient
- Skin grafting for chronic leg ulcers
- Hip replacement or other orthopaedic procedures for avascular necrosis
- Surgical drainage of the penile corpora for resistant priapism
- Insertion of a penile prosthesis if impotence occurs
- Cholecystectomy for gallstones (whether acute cholecystitis is present or not)
- Treatment of stroke in SCD
- Admit
- Pharmacologic management of cerebral edema and seizures (avoid fluid overload)
- Surgical repair of ruptured aneurysms
- Emergency exchange transfusion (reduce HbS < 30% of total hemoglobin, stop occlusion and sickling)
- Treatment of ACS in SCD
- Admit
- Oxygen
- Antibiotics
- IV hydration (avoid volume overload)
- Pain management
- Empiric broad spectrum antibiotics
- Bronchodilator therapy (if reactive airway disease in children)
- Emergency exchange transfusion (for severe disease)
- Long-term hydroxyurea (to reduce recurrence)
- Treatment of Acute Multi-Organ System Failure in SCD suspect if there is typical vaso-occlusive pain that is followed with deterioration in about 3-4 days of admission
- Pain management
- Hydration
- Oxygenation
- Blood Cultures + empiric broad-spectrum antibiotics
- Monitor electrolytes and fluids
- Intubate if needed
- Dialysis if needed
- Aggressive tranfusion (simple or exchange) if Hb is dropping rapidly – target 10g/dL.