Overview
Hemolytic anemia is simply anemia due to the destruction of red blood cells. The destruction of red blood cells happens at an accelerated pace, beyond the capacity of the bone marrow to compensate. Usually, a normocytic anemia is present unless there is another co-existing anemia. Patients commonly present with symptoms of anemia with no other features. In addition to getting an iron panel, serum LDH, haptoglobin and total bilirubin should be obtained. LDH is elevated and haptoglobin is reduced in hemolytic anemia. Reticulocytes will be elevated (>1.5%) in normal bone marrow response.
Summary
| Association | Coombs | Other tests | Treatment | |
|---|---|---|---|---|
| Warm AIHA | Autoimmune condition, Lymphoma | Positive | – | Corticosteroids |
| Cold AIHA | Infection and exposure to cold | Positive | Cold agglutinin titer | Avoid cold |
| Drug-induced AIHA | Drug exposure (cephalosporin #1) | Variable | – | Remove offending drug |
| Hereditary spherocytosis | Splenomegaly and FHx | Negative | Spherocytes in PBF | Splenectomy if severe |
| PNH | Cola-coloured urine in the morning +/- pancytopenia, thrombocytopenia | Negative | Flow cytometry for CD55/59 | Eculizumab |
| G6PD deficiency | Male, Intermittent hemolysis following oxidant | Negative | Heinz bodies and bite cells in PBF | Avoid oxidants |
Classification of hemolytic anemia
| Intrinsic hemolytic anemia | Membranopathy, Enzymopathy, Hemoglobinopathy |
|---|---|
| Extrinsic hemolytic anemia | Autoimmune and alloimmune hemolysis, Mechanical destruction, Toxin-mediated destruction, Hypersplenism |
Mechanism of RBC destruction
| Intravascular hemolysis | Hemolysis in blood vessels |
|---|---|
| Extravascular hemolysis | Hemolysis that occurs in the spleen or liver |
Coombs positive vs Coombs negative hemolytic anemia
| Coombs (+) | Warm and Cold autoimmune hemolytic anemia, Drug-induced hemolytic anemia |
|---|---|
| Coombs (-) | Hereditary spherocytosis, Paroxysmal Hemoglobinuria, Glucose-6-Phosphate deficiency, Sickle cell disease |
- Signs and symptoms
- Fatigue, weakness, pallor, breathlessness
- Splenomegaly (due to trapping and breakdown of RBCs in the spleen)
- Jaundice, Icterus, and Pruritus (due to degradation of hemoglobin into bilirubin)
- Gallstones (due to hyperbilirubinemia)
- Hemosiderinuria (due to destruction of RBCs)
- Constitutional symptoms – fever, weakness, chills (primarily in acquired hemolytic anemia)
- Investigations
- Complete blood count
- Decreased RBC count
- Decreased Hb
- Decreased HCT
- Normal MCV
- Elevated WBC
- Decreased Platelets in MAHA
- Reticulocyte count
- Elevated reticulocyte count
- Peripheral blood film
- Reticulocytosis Intravascular hemolysis
- Heinz bodies (G6PD deficiency)
- Schistocytes (MAHA and macroangiopathic haemolytic anemia)
- Intracellular organisms (maleria, babesiosis, bartonellosis)
- Spherocytes (Hereditary spherocytosis and immune-mediated hemolysis)
- RBC agglutination in CAD
- Sickle cells in sickle cell disease
- Target cells in sickle cell disease, thalassemia, HbC disease
- Teardrop cells in thalassemia
- Hb Crystals in HbC disease
- Smudge cells (Gumprecht shadows) in CLL
- Reticulocytosis Intravascular hemolysis
- Serum studies
- Decreased haptoglobin
- Increased LDH
- Elevated indirect bilirubin
- Urinalysis
- Haemoglobinuria
- Hemosiderinuria
- Urobilinogen
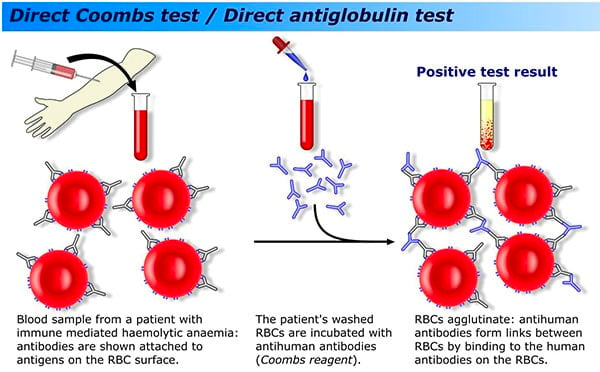
- Coombs test
- Positive Coombs test: Autoimmune process
- Complete blood count
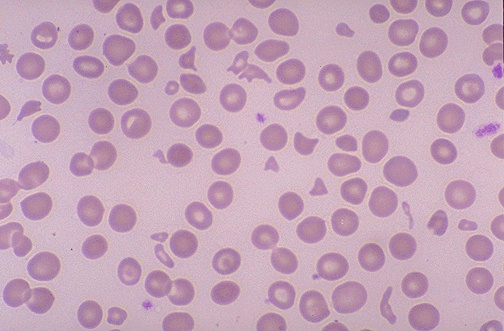
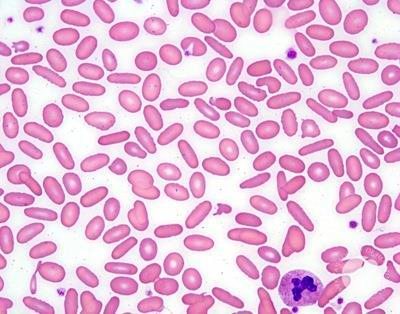
Warm-Agglutinin Hemolytic Anemia
Warm coombs (+) hemolytic anemia is usually idiopathic. It can be linked to a lymphoproliferative disorder or autoimmune disease. Patient produce IgG against their own RBCs. The tagged RBCs are destroyed in the spleen (extravascular hemolysis)
- Causes of autoimmune hemolytic anemia
- Idiopathic
- Lymphoproliferative disease: CLL, Lymphoma
- Autoimmune disease: SLE, scleroderma
- Investigations
- Coombs test: positive
- Cold agglutinin titer: negative
- Treatment
- Folate supplementation
- Corticosteroids e.g. Prednisone: best initial step
- Rituximab
Cold-Agglutinin Hemolytic Anemia
Coombs (+) hemolytic anemia is usually linked to infection. Classically caused by Mycoplasma, EBV, HIV. Rarely associated with CLL or Lymphoma. The patient produces IgM molecules which activate complement leading to intravascular hemolysis.
- Patient History
- History of “walking pneumonia” – fatigue, breathlessness, cough
- History of mononucleosis
- History of lymphoma
- Signs and symptoms
- Worsened with exposure to cold
- Purplish discoloration of the fingers and toes
- Investigations
- Coombs test: positive
- Cold-agglutinin titer: Positive
- Treatment
- Folate supplementation
- Avoid cold conditions
- Rituximab (can be used but not necessary)
Drug-Induced Hemolytic Anemia
Coomb (+) hemolytic anemia linked to a drug
- Causes of drug-induced hemolytic anemia
- Cephalosporins (most common)
- Levofloxacin
- Nitrofurantoin (UTI)
- Rifampin (anti-TB)
- Carbidopa or Levodopa (Parkinson’s disease)
- alpha methyldopa (hypertension in pregnancy)
- Investigations
- Coombs: Positive
- Treatment
- Stop the offending drug
- Folate supplementaiton
- Steroids may be effective if Coombs (+)
Hereditary spherocytosis
An Autosomal Dominant condition where RBCs lack spectrin and lose their flexibility and stability. These red blood cells are destroyed in the spleen.
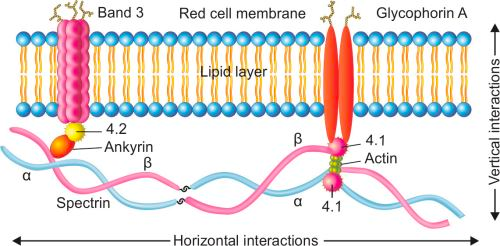
- Red cell membranopathies
- Disordered Vertical interactions
- Hereditary spherocytosis
- Disordered Horizontal interactions
- Hereditary elliptocytosis
- Hereditary pyropoikilocytosis
- South East Asian ovalocytosis
- Disordered Vertical interactions
- Etiology
- Genetic mutations in proteins that have vertical interactions (Ankyrin, Spectrin, Band 3)
- Pathogenesis
- Defects in RBC membrane proteins (Ankyrin or spectrin) responsible for tying the inner membrane skeleton with the outer lipid bilayer
- Continuous loss of lipid bilayer components as microvesicles
- Decrease membrane surface area (microspherocyte) in relation to volume
- Microspherocytes with decreased membrane stability
- Inability to deform while going through narrowed vessels
- Splenomegaly: Entrapment within splenic vasculature (Erythrostasis)
- Extravascular hemolysis: Destruction by splenic macrophages
- Signs and symptoms
- Pallor, fatigue, breathlessness
- Jaundice; Marked if associated with Gilbert’s disease (Increased unconjugated bilirubin)
- Splenomegaly (Erythrostasis)
- Black pigment gall stones (High levels of bilirubin)
- Aplastic crisis (precipitated by parvovirus infection)
- Investigations
- Complete blood count
- Low Hb
- Low PCV
- Low MCV
- Increased MCHC
- Reticulocytes:
- 5 – 20%
- Peripheral blood film
- Microspherocytes
- Osmotic fragility test: best initial test
- Increased
- Direct Coomb’s test: Negative (Rule out auto-immune hemolysis and hemolytic transfusion reactions) ****
- **Rapid fluorescent flow analysis of eosin (**Malemide bound to red cells, used as a test for HS and membrane band 3 protein deficiency
- Rx: Splenectomy
- Complete blood count
- Osmotic fragility test
- Osmotic fragility test is used to measure RBC resistance to hemolysis when exposed to a series of increasingly dilute saline solutions (the sooner the hemolysis, the greater the osmotic fragility)
- Factors affecting the osmotic fragility test
- Cell membrane permeability
- Surface area-to-volume ration
- Differentials for a positive osmotic fragility test
- Hereditary spherocytosis
- Autoimmune hemolytic anemia
- Hemolytic transfusion reactions
- Older RBCs are also more fragile
- Hemolytic anemia
- Treatment
- Folate replacement
- Elective splenectomy (to stop hemolysis and hyperbilirubinemia)
- What are the characteristics of microspherocytes?
- Small
- Dark
- Very round
- Hyperchromatic
- No central pallor
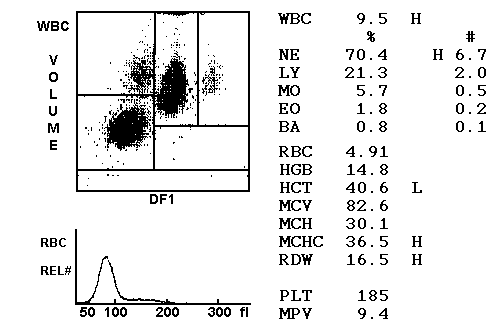
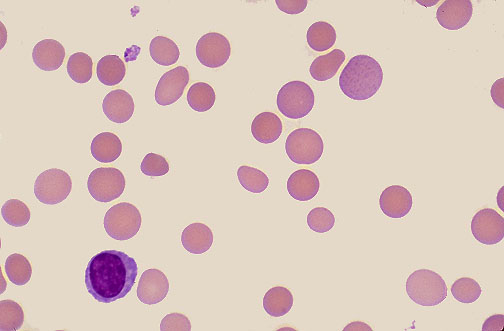
Glucose-6-Phosphate Dehydrogenase Deficiency
G6PD deficiency is and X-linked recessive condition that yields a reduction in the antioxidant NADPH. Often presents acutely in childhood and is linked to infection, oxidative agents, and foods.
It is the most common human enzyme deficiency. Prevalence is 400 million world-wide. Prevalent in Black, Asian, Middle Eastern and Mediterranean individuals.
WHO classification of G6PD variants
| Class I | Severe enzyme deficiency | Chronic hemolysis |
|---|---|---|
| Class II | Severe enzyme deficiency (1-10% residual activity) | Intermittent acute hemolysis |
| Class III | Moderate enzyme deficiency (10-60% residual activity) | Intermittent acute hemolysis |
| Class IV | No enzyme deficiency (60-150% activity) | |
| Class V | Increased enzymatic activity (>150%) |
- Causes of acute hemolysis in G6PD deficiency
- Infections
- Acute illness e.g. Diabetic Ketoacidosis
- Fava beans
- Henna
- Drugs such as:
- Antimalarial drugs: chloroquine, primaquine
- Sulfa drugs – TMP-SMX
- Nitrofurantoin
- Isoniazid
- Dapsone
- NSAIDs
- Ciprofloxacin
- Chloramphenicol
- Pathogenesis
- G6PD is the rate-limiting enzyme of HMP
- HMP yields NADPH
- NADPH is essential to convert oxidized glutathionine back to reduced glutathione
- Reduced glutathione is capable of neutralizing ROS and free radicals therefore protecting RBCs from oxidative damage
- G6PD deficiency
- Reduced glutathione
- Red blood cells become susceptible to oxidative stress (infections, acute illness, food, drugs) that can damage erythrocyte membrane
- Results in intravascular and extravascular hemolysis
- Patient History
- Male
- African, Middle-Easter, Asian or Meditterenean descent
- Hx of oxidant
- Hx of infection or acute illness
- Signs and symptoms
- Most are asymptomatic
- Recurring hemolytic crises (Acute hemolytic anemia) in response to oxidative stress
- Sudden onset of back or abdominal pain
- Jaundice
- Dark urine (due to hematuria or hemoglobinuria
- Transient splenomegaly
- Recurrent severe infections causing symptoms of chronic granulomatous disease
- Investigations
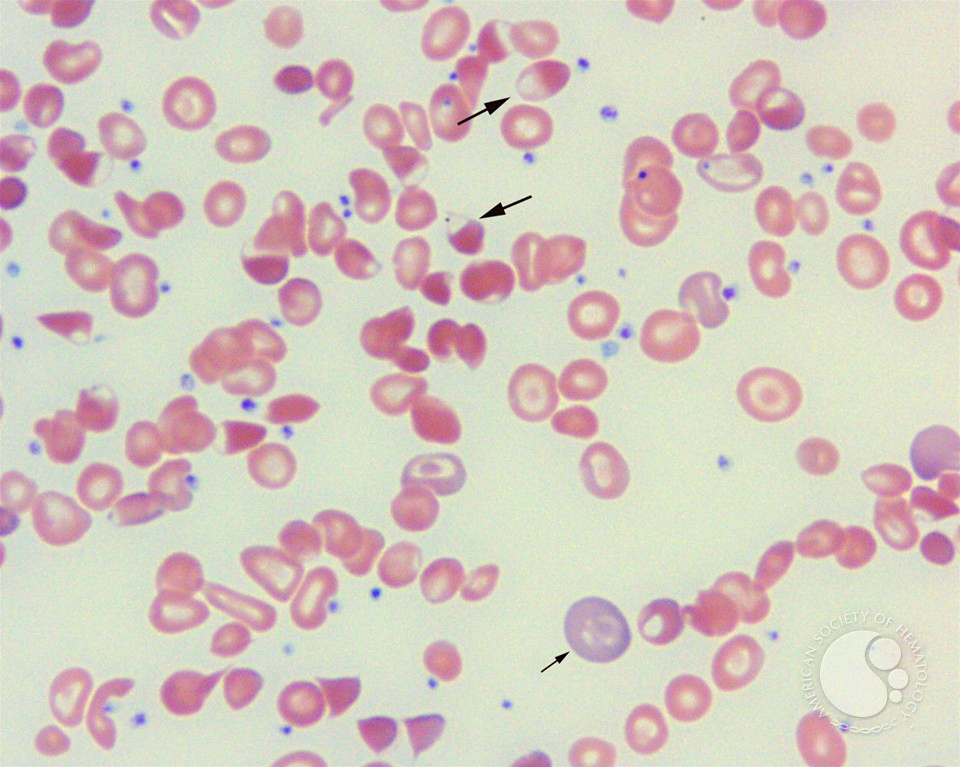
- Peripheral blood film
- Contracted and fragmented cells
- Heinz bodies (Oxidative stress causes Hb denaturation and precipitation which appear as purple spots)
- Bite cell and Blister cells (Heinz bodies removed by spleen)
- Signs of intravascular hemolysis
- Normocytic anemia
- Increased reticulocyte count
- Increased unconjugated bilirubin
- Increased LDH
- Decreased haptoglobin
- Hemoglobinuria
- **Screening: Fluorescent spot test (**Qualitative measurement of G6PD enzymatic activity)
- Confirmatory: Quantitative G6PD enzyme analysis
- Molecular test: Detection of G6PD gene mutation
- Peripheral blood film
- Treatment
- Stop offending drug or food
- Treat infection or illness
- Hydration in case of severe hemolysis
- Phototherapy or exchange transfusion if neonatal jaundice
- Blood transfusion (only in rare, severe cases)
- Educate the patient
- Methemoglobin reduction test
- Rapid indirect test that measures reduced methemoglobin levels produced after NADPH oxidation
- G6PD activity is assessed by first treating RBC with nitrite
- Oxyhemoglobin (red) is converted to methemoglobin (brown)
- The rate of NADPH-dependent methemoglobin reduction is then examined in the presence of an appropriate redox catalyst (Nile blue or methylene blue) and substrate (glucose)
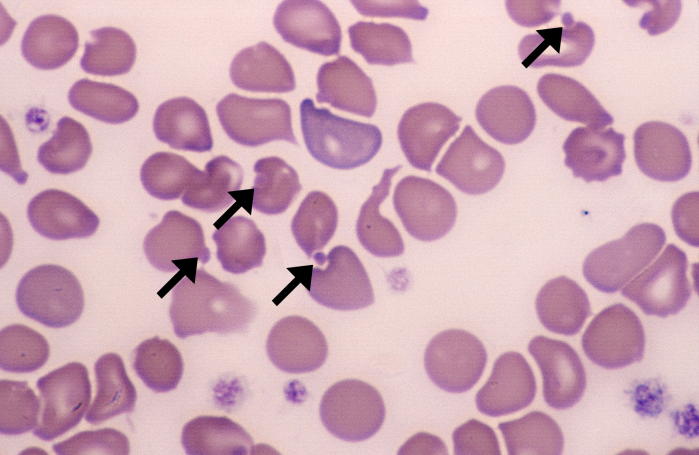

Paroxysmal Nocturnal Hemoglobinuria (PNH)
PNH is an idiopathic condition due to clonal defects in the RBC membrane protein (GPI) which is responsible for inactivating complement. There is increased activation of complement and intravascular hemolysis. Patients with PNH have an increased risk of thrombosis (hepatic thrombosis, dermal thrombosis).
- Why does PNH occur at night?
- Compliment activation is enhanced by the slightly acidotic state that happens during sleep
- Pathophysiology
- Acquired mutation on the PIGA gene located on X chromosome
- Deficiency in CD55/DAF (Decay-accelerating factors) and CD59/MIRL (Membrane Inhibitor of reactive lysis) which are part of the GPI anchor protein
- GPI defect leads to increased activation of complement leading to intravascular hemolsysis
- GPI defect in WBCs and Platelets → Pancytopenia
- Complement activation in platelets → activation → increased risk of thrombosis
- Signs and symptoms
- Fatigue, pallor, breathlessness
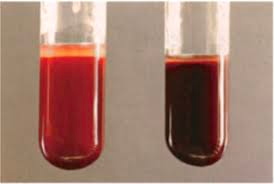
- Dark “cola” or “tea-coloured urine” when waking up
- Investigations
- Flow cytometry: most accurate test
- Deficiency in CD55/59
- Acidified serum lysis “Ham” test
- Sugar-water test
- Urinalysis
- Hemoglobinuria
- Flow cytometry: most accurate test
- Treatment
- Eculizumab: first-line
- Iron and Folate supplementation (iron supplementation since hemolysis is intravascular and iron is lost in urine)
- Steroids if severe
- Anticoagulation
- Heparin is mandatory if pregnant
Pyruvate Kinase Deficiency
Autosomal recessive condition caused by deficiency of pyruvate kinase.
- Pathophysiology
- Glucose is the only source of energy for RBC
- Pyruvate Kinase catalyses the last step of glycolysis (conversion of PEP to pyruvate)
- The bsence of pyruvate kinase
- ATP is deficient in RBC s
- ATP deficiency disrupts cation gradient along RBC membrane
- Rigid RBCs
- Increased hemolysis (Extravascular)
- Why are there masked symptoms of anemia in pyuvate kinase deficiency?
- Accumulation of 2,3-BPG causes O2 release from Hb
- Signs and symptoms
- Jaundice (typically newborn due to hemolysis and history of exchange transfusions)
- Gall stones
- Frontal bossing
- Splenomegaly
- Pallor, fatigue, weakness
- Hydrops fetalis: rare cases
- Investigations
- Poikilocytosis and distorted prickle (Burr or echinocytes) cells
- Decreased pyruvate kinase enzyme activity
- PKLR gene mutation
Other Membranopathies
- Hereditary Elliptocytosis
- Autosomal dominant
- Homozygous patients present with severe hemolytic anemia
- Mutations in proteins involved in horizontal linkages in the membrane skeleton (spectrin and protein 4.1
- Defective spectrin dimer formation
- Defective spectrin-ankyrin associations
- Protein 4.1 deficiency or abnormality
- South Asian Ovalocytosis
- Nine amino acid deletion at the junction of the cytoplasmic and transmembrane domains of the band 3 protein